

Automatic Genome Annotation
HINWEIS: Der Vortrag wird in englischer Sprache gehalten!
In part of the talk I will give an overview over approaches to structural genome annotation including approaches based on RNA-Seq and approaches exploiting homology.
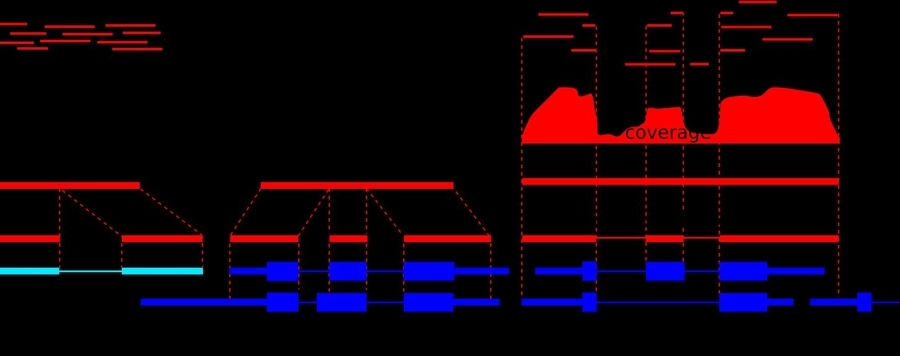
Our group is developing the gene finder AUGUSTUS. In particular we currently work on methods for the 'clade annotation problem'. We aim to simultaneously annotate a set of aligned genomes, e.g. 16 newly de novo assembled mouse strains, several hundred strains of Staphylococcus aureus (pan-genome annotation) or more diverse sets of vertebrate genomes.
Some loci in some species may already have a trustable annotation, some genomes may have evidence, e.g. from RNA-Seq, some genomes may be naked.
The multiple alignment of genomes can be used to transfer evidence across species and also provides strong evolutionary evidence through negative selection and sequence conservation.
The new version of our gene finder AUGUSTUS addresses the simultaneous identification of the structure of all genes in all aligned genomes, thereby respecting structural differences but exploiting synteny, conservation and negative selection.
Our model leads to an NP-hard optimization problem on a graph, that we can nevertheless often solve exactly and in acceptable time.
Ansprechpartnerin / Ansprechpartner


