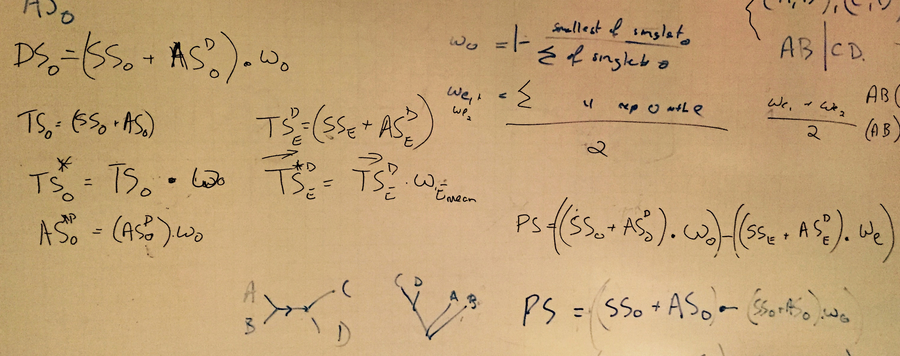Quick facts
Project title:
Countering Systematic Bias in Phylogenetics through Non-Parametric Analyses of Quartets
Description
The assessment of phylogenetic relationships provides a foundation for the interpretation of all
comparative biological data and thus phylogenetic tree reconstruction a central task of modern biology. Recent phylogenomic analyses comprising hundreds of genes and taxa offers, for the first time, the opportunity to investigate all changes in complete genomes by simultaneously reducing stochastic sampling errors. So the goal of approximating the “Tree of Life” seems to be more attainable today than ever. However, the phylogenomic era seems to be also the beginning of an era of strong incongruence due to the further accumulation of results affected by systematic biases arising from rate or compositional heterogeneity from insufficient substitution models or insufficient detection of phylogenetic signal, which can lead to strongly supported but incorrectly resolved phylogenetic relationships.
An important source of systematic bias and probably the most frequently cited reason for
incorrect placement of taxa in phylogenetic reconstructions is surely long branch attraction (LBA). LBA can be described as inherent bias due to a combination of long and short evolutionary paths, in which random similarity based on convergent or parallel character changes lead to an artifactual phylogenetic grouping. Recent simulation studies show that even a slight model misspecification can cause incorrect topologies in probabilistic based analyses (Kück et al. 2012: long branch effects distort Maximum Likelihood phylogenies despite selection of the correct model; Kück & Wägele 2015: Plesiomorphic character states cause systematic errors in molecular phylogenetic analyses: a simulation study).
One likely reason for misspecifications in modern probabilistic substitution models is the usual assumption of time reversibility. The direction of character evolution along a tree is not considered by these models and therefore actual probabilistic based tree analyses do not incorporate an important step of Hennigian phylogenetic inference, the distinction between new (apomorphic) and old (plesiomorphic) homologies (Kück & Wägele 2015: Plesiomorphic character states cause systematic errors in molecular phylogenetic analyses: a simulation study).
The project is focused on the developing of PhyQuart, a new quartet-based algorithm which considers two alternative directions of character evolution along the internal branch of a quartet tree to discern between potentially apomorphic and plesiomorphic split-supporting site-patterns, and ML to estimate the expected number of convergent split-supporting site-patterns. This combination of Hennigian logic and ML estimation represents a completely new strategy for the evaluation of sequence data.
The efficiency of the new approach in detecting phylogenetically informative and conflicting signals in 4-taxon cases has been already successfully tested through extensive quartet simulations, including cases with strong branch length differences. Their results show that the combination of site-pattern and ML analyses in PhyQuart leads to quartet inferences that especially in cases of strong branch length heterogeneity are better than in conventional Maximum Likelihood analyses.
Lowest reconstruction success observed for different branch length heterogeneties (red branches are stepwise elongated, x-axis) and alpha scores given 100 quartet simulations (y-axis) for each branch length condition using ML versus PhyQuart.
A formal description of the algorithm and a complete overview of all conducted performance tests based on simulated data are given in ''Kück et al. 2017: Can quartet analyses combining maximum likelihood estimation and Hennigian logic overcome long branch attraction in phylogenomic sequence data?''. The current manuscript is primarily designed to address a general description of the (currently quartet limited) PhyQuart algorithm with insights about its efficiency and robustness in identifying phylogenetic signal strengths compared to conventional maximum likelihood analyses in cases of different branch length heterogeneities of simulated quartets. This article is just the beginning of a longer, promising, hopefully fruitful journey of continuous algorithmic development and improvement towards a finally robust and efficient supertree method.
The algorithm introduced in this study is implemented in a new software tool called PENGUIN, a command line driven PERL script that runs on Windows PCs, Mac OS and Linux operating systems and can be easily implemented into automatic process pipelines. PENGUIN is freely available and released under the terms of the GNU General Public License (GPL) 3.0. The software script as well as the corresponding manual and example files can be downloaded from https://github.com/PatrickKueck/Penguin.
Overview of analytic capabilities of the actual PENGUIN software script.
Yet, the PENGUIN software allows the analysis of all quartets of terminal taxa or clans in larger trees and therefore provides a new tool to detect phylogenetic support and contradicting signals which can be used to assess the robustness of hypothesized clan relationships, named PhyQuart-Mapping, a priori tree reconstruction or within an already existing, more complex tree. In a new study (Kück et al. submitted) based on simulated and empirical data we show that PhyQuart-Mapping signals conflict in the data where other tools support wrong topologies. The PENGUIN software may also be useful for identifying individual rogue taxa that are difficult to place due to ambiguous or weak phylogenetic signal. This characteristic of rogue taxa should become visible when multiple quartets selected from predefined multi-taxon clans are analysed.
Future plans of the project comprise (among algorithmic improvement of PhyQuart) inter alia to improve the selection of highly informative and thus appropriate quartets (e.g. quartet topologies without much signal conflict) in combination with quartet-based supertree methods as well as for the development of networks summarising conflicting signal and providing information on the probable location of the root in trees or networks independent of any consideration of outgroups.
Location