

COGNATE
COGNATE: Comparative Gene Annotation characterizer

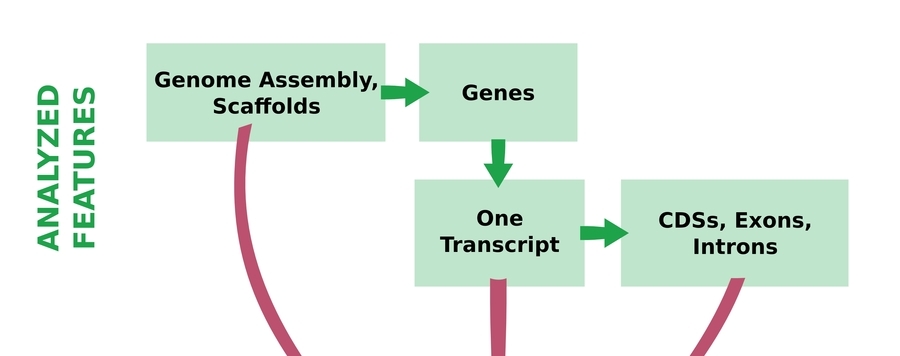
COGNATE is a tool to simultaneously analyze a given protein-coding gene annotation and the corresponding assembled sequences of a genome. It allows a quick and easy extraction of basic genome feature and gene repertoire data. Thus, it is a tool to primarily describe a genome and its annotation of protein-coding genes, which is an essential prerequisite for comparative and meta-analyses of genome and gene structure.
COGNATE infers the following main parameters:
- summary counts of the analyzed features
-
strandedness of transcripts and their CDSs, exons, and introns
- length statistics (nucleotide sequences/amino acid sequences), including N50/L50, 75/L75, N90/L90
- intron length distribution as suggested by [1]
-
percental GC content statistics in two different ways, namely
- using a calculation that explicitly considers IUPAC ambiguity codes (G, C, S per total length excluding N, R, Y, K, M, B, D, H, V)
- using the previously prevailing calculation of GC per total length, which is inappropriate for genome comparisons due to its dependence on assembly quality
- statistics of CpG dinucleotide depletion (CpG observed/expected), normalized by C and G content of the respective region [2]
- density statistics (ratio of the length of a feature covered by another, number-wise)
- coverage statistics (ratio of the length of a feature covered by another, length-wise).
The output design is focused on clarity and the combination of overview display and detailed parameter evaluation to make COGNATE as useful as possible within its designated scope.
Usage of the tool COGNATE requires a working perl installation and GAL [3], which is originally provided by the Sequence Ontology [4], written by Barry Moore. The GAL/lib/ directory is included with minor changes as GAL/ in the distribution of COGNATE and thus requires no further installation.
For bug reports, feature requests, and older versions, please contact Jeanne Wilbrandt or Bernhard Misof.
Cite as
Wilbrandt J, Misof B, Niehuis O (2017) COGNATE: Comparative Gene Annotation characterizator. BMC Genomics 18:535.
https://github.com/ZFMK/COGNATE
References
-
Roy SW, Penny D. Intron length distributions and gene prediction. Nucl. Acids Res. 2007;35:4737–42.
-
Elango N, Hunt BG, Goodisman MAD, Yi SV. DNA methylation is widespread and associated with differential gene expression in castes of the honeybee, Apis mellifera. PNAS. 2009;106:11206–11.
-
Genome Annotation Library, https://github.com/The-Sequence-Ontology/GAL
-
Eilbeck K, Lewis SE, Mungall CJ, Yandell M, Stein L, Durbin R, et al. The Sequence Ontology: a tool for the unification of genome annotations. Genome Biology. 2005;6:R44.
Figure
Contact person

Documents

