

BaCoCa
BaCoCa (Base Composition Calculator)
BaCoCa is designed to perform multiple statistical analyses on multiple nucleotide and amino-acid sequence alignments.
The results of the BaCoCa analyses can be used for a detailed and statistical comprehensive data evaluation.
Furthermore, the results can help to identify phylogenetic sequence biases which can lead to incorrect tree reconstructions.
The program can handle hundreds of user specified gene and taxon partitions of a single sequence input file in one process run.
BaCoCa is a command-line driven program written in Perl and works on WindowsPCs, Macs and Linux running systems.
Therefore, it can be easily integrated into automatic pipeline processes of phylogenomic studies.
All results issued by BaCoCa can be directly integrated into further analyses using statistical R packages.
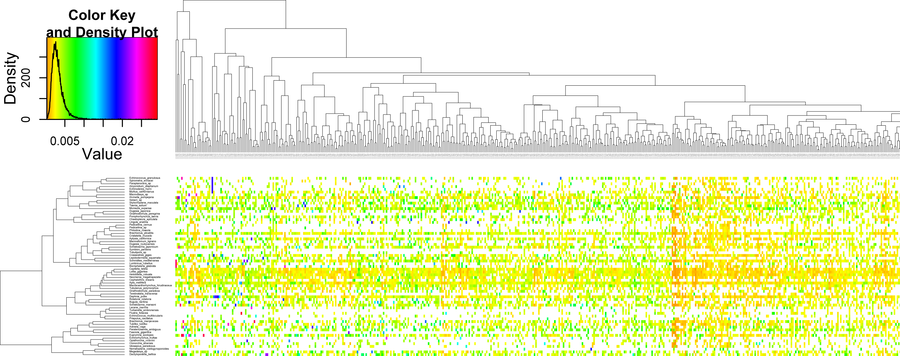
For example, heat map analyses of taxon versus gene matrices can be used to find clusters of genes and/or taxa with similar properties.
Furthermore, all calculations of the BaCoCa software program are very fast and can be easily executed on a normal desktop computer, even if data sets consist of phylogenomic data.
The downloadable BaCoCa.zip file contains the BaCoCa executable Perlscript (BaCoCa.v1.0beta.pl), a detailed documentation of all BaCoCa implemented calculations as well as detailed information of usage and BaCoCa result outputfiles (BaCoCa_Manual.pdf), and example infiles of empirical nucleotide (BaCoCa_Example_Files_NUC) and amino acid (BaCoCa_Example_Files_AA) supermatrices.

Example heat map of a BaCoCa compositional heterogeneity result file (generated with R) of 559 genes and 65 taxa
Contact person


